AntiBase
A Data Base for the Identification AntiBase can be ordered directly from the author or from Wiley Interscience. A full Scidex version of AntiBase 2011 can be downloaded from the following link: A valid key code for activation can be purchased from the author. Updates are available (presently) once a year; for prices, see the flyer A preliminary manual
and a test version (use alternatively the following link) can also be downloaded here. The activation code for the free
test version is 455603568.
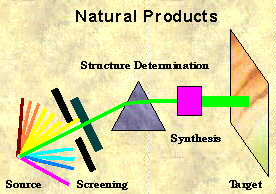
Progress in natural products chemistry today is progress in synthetic chemistry tomorrow! Structure determination of complex natural products delivers not only the most interesting, the
most challenging targets for synthetic chemists, but is also the basis of new medical applications of the future. There is no doubt that natural product's isolation and structure identification is still
one of the most important areas in chemistry. An army of scientists tries to discover with unlimited effort a limited number of natural
metabolites: More than 170.000 natural products are known today, and every year are added about 700 new structures just only from micro-organisms. This gives raise to a major problem in
natural product chemistry: It is unavoidable that certain common compounds are re-isolated again and again, a handicap which costs time and money, and is a steady source of avoidable frustration. The chemical recognition of doublets proceeds usually by comparison of experimental results with
data from literature or own experience as a sequence of logical yes/no decisions. This process is normally restricted by the limited capacity of the brain and can therefore be done much better by
a computer, if a suitable database is available. The number of chemical databases is sufficiently high, the most important being Chemical
Abstracts and the Beilstein file. Smaller ones provide just only one set of data for each compound as those delivered as part of spectroscopic equipment (IR, UV, MS). Also bigger ones may not
contain chemical structures (Berdy's database) or are restricted mainly to medical relations (NAPRALERT) and are not suitable in every respect for structure determinations.
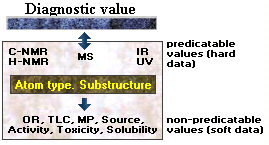
Besides to search chemical structures and to do substructure searches, a "suitable" database should provide predominantly hard facts, i.e. data which is connected directly with chemical
structures, and vice versa. The most important information are certainly 13C and 1H NMR-spectra, mass spectra and - with decreasing importance - UV and IR data. Soft data like
melting points, optical rotations, Rf values or biological activities do not have obvious correlation to chemical structures. The Dictionary of Natural Products database (Chapman & Hall) and AntiBase (Chemical
Concepts) are especially useful for structure determination; a third one (Crossfire from Beilstein) may do the same in near future. The Dictionary of Natural Products covers all natural products,
but has - compared with AntiBase - slower search capabilities and only very limited spectral information. AntiBase is delivered together with the data managing system Scidex. Its data set covers mostly
compounds from micro-organisms and higher fungi, including yeasts, ascomycetes, basidiomycetes and lichens, but also algae and cyanobacteria (to date more than 32.000
compounds). Due to the use of Scidex, a full range of structure and substructure search capabilities is available. The most important feature of AntiBase is the fact that for nearly all
metabolites with known structures, either experimental or SpecInfo-calculated 13C NMR data are available. In the new Scidex version, these data can be searched also structure-sensitive: Even a
search by the number of quaternary C atoms, which is easily determined from 13C spectra, or the number of carbonyl, methin or methylen groups is possible. Also coupling patterns or signal shifts
and multiplicities from 1H NMR spectra can usually be translated into sub-structures: A typical search procedure is described for AntiBase, using mainly its spectral information: The 13
C NMR spectrum of an unknown antibiotic shows 28 signals. The search for compounds with 27-29 carbon atoms gave 1125 answers out of 12.700 entries with calculated or experimental 13C
NMR data; 415 of them contained an aromatic and/or heterocyclic ring, which was indicated by signals between 100-140 ppm. 240 compounds of this answer set contained at least one carbon
atom at 170-175 ppm, 33 no other signals between 150-250 ppm. The search in this group for metabolites with exactly two methylen groups gave only seven answers: all of them were
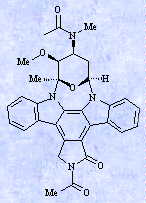
staurosporin derivatives! The metabolite was easily determined as staurosporine by comparison of the residual 13C values. This answer - staurosporine - was also obtained by a search for aromatic compounds with 27-29 C
atoms, exactly one carbonyl group, even or more quaternary carbon atoms, two CH2 and three CH3 groups. The same result can be obtained by various other search strategies and their combinations. Search strategies for AntiBase. + indicates presence, - absence of the search term. One of the most remarkable features of AntiBase is its capability in peptide identification. As also
amino acid fragments can be used for a sub-structure search, the result of an amino acid determination after hydrolysis of a peptide and determination of the N-terminal acid usually is
sufficient to evaluate unambiguously most of the more than 1500 peptides in the data base. For various reasons only in few cases, complete data sets are available for natural products. But
as practice shows, even overlapping of fragmentary information is sufficient for a concise determination in most cases: Due to its spectroscopic information, AntiBase is also a versatile tool for the structure
determination of hitherto unknown compounds, as the staurosporin example may already have shown: As nearly every new compounds has already known relatives already in the database, a
search will list up easily variations with the same chromophor or with similar shifts. Recognition of re-isolated known compounds is much easier now as without computer-assisted
knowledge bases. But these are still far away from being perfect: 13C NMR data can be searched easily by their shift, but even for 1H NMR spectra of natural products precise values - exact
shifts and coupling constants of complex spin patterns - are lacking in most cases in the literature. Mass spectra are very often not reproducible and usually are not reported for the complete range
of signals, the presentation of IR spectra by a selection of only a few values is finally completely useless for identification purposes. The valuable and very often irreplaceable information of the
original spectra remains unused or even gets lost. Only a computer-searchable spectrum or at least a figure of good quality might be a way out.
Unfortunately, most scientific journals cannot provide the space to print the spectra. It is strongly suggested therefore to collect the spectra of at least all natural products in a computer-readable
format with sufficient chemical intelligence on a central place. This should be done on a national or better international level by a central non-commercial organisation, where all chemists can
have access via STN or internet. This is certainly nothing that can be done immediately, but in the meanwhile spectra could be kept and processed by Chemical Concepts1)
, the AntiBase maintenance group2) or perhaps also institutions like the Beilstein. But we should never forget that all this can have success only if the
necessity for a co-operation is widely accepted and work is fully supported by every chemist nationwide! The database of the future might then be able to sort out unwanted constituents online
during the separations to save more time for the real problems. |

Antibiotics and other Fermentation products for affordable prices
Staurosporine, cryst., NMR-spetroscopally pure: 10 mg vials each 3000.- Euro Prices for Actinomycins, Valinomycin, Reductiomycin, ß-Indomycinone and other antibiotics on request
1 mg vials each 350.- Euro
0.5 mg vials each 190.- Euro
0.1 mg vials each 40.- Euro